
Doctor Yohann Grondin
Facultad de Ciencias Biológicas Centro de Investigación y Desarrollo en Ciencias de la Salud / UANL
Doctora Rocío Ortiz López
Departamento de Bioquímica y Medicina Molecular Facultad de Medicina Centro de Investigación y Desarrollo en Ciencias de la Salud / UANL
anidemsolrac@yahoo.com
 Las ciencias ómicas son un grupo de disciplinas relativamente nuevas, que empezaron a emerger hace una década. Las primeras, basadas en el Dogma Central de la Biología, fueron la Genómica, la Transcriptómica y la Proteómica, las cuales son ya muy conocidas y muy bien definidas. La primera se enfoca a la determinación y mapeo del grupo de genes de diferentes organismos; la segunda, al estudio de la expresión de los ácidos ribonucleicos mensajeros (RNAm), y la tercera, al estudio de la estructura y función de las proteínas.
Las ciencias ómicas son un grupo de disciplinas relativamente nuevas, que empezaron a emerger hace una década. Las primeras, basadas en el Dogma Central de la Biología, fueron la Genómica, la Transcriptómica y la Proteómica, las cuales son ya muy conocidas y muy bien definidas. La primera se enfoca a la determinación y mapeo del grupo de genes de diferentes organismos; la segunda, al estudio de la expresión de los ácidos ribonucleicos mensajeros (RNAm), y la tercera, al estudio de la estructura y función de las proteínas.
Algunas otras de estas ciencias forman una lista creciente y exhaustiva, pero cada una de ellas va emergiendo por su importancia, como la Metabolómica, que se refiere al análisis integral de los metabolitos celulares. La Farmacogenómica analiza el efecto que tienen algunas variaciones genómicas, como los polimorfismos de nucleótido simple (SNPs, en inglés) sobre la cinética y la dinámica celular y tisular de la respuesta a diferentes medicamentos.
También hay varias otras “ÓMICAS” menos evidentes y definidas, como la ORFeómica, una nueva disciplina que intenta amplificar, clonar y evaluar todos los marcos de lectura abierta [open reading frames (ORFs)] descubiertos en cada genoma, que pudieran predecir nuevas proteínas codificantes. La Glicómica busca estudiar los azúcares y sus combinaciones en un organismo, y el papel que ellos juegan.
NUEVOS NOMBRES
Sin profundizar en más detalles sobre el resto de disciplinas existentes y algunas otras que están por emerger, podemos especular que algunos de los grandes nombres que oiremos en los próximos años van a ser los de:
Interferómica, relacionada con el estudio de los RNAs de interferencia de cada organismo (y que en el humano son muy promisorios para el diseño de medicamentos), o inclusive con la interacción entre ellas, como la Histonómica con la Metilonómica, relacionadas con la caracterización e identificación de los códigos asociados a los cambios en la regulación epigenética; es decir, los cambios que ocurren en el DNA y la cromatina después de la replicación del material genético.
Debido a que las tecnologías genómicas se han movido de secuenciar un gen a secuenciar todo el genoma, ahora es posible secuenciar, en un solo ensayo, los genomas de microorganismos de un medio ambiente (Metagenómica). El producto de estos datos de metagenómica, sin embargo, genera otro reto para su manejo, debido a que las secuencias no siempre están asociadas con especies específicas, como había sido tradicionalmente conceptualizado (un gen en un solo genoma); ahora deben considerarse las asociaciones entre los diferentes microorganismos y sus variantes en diferentes proporciones.
MILLONES DE SECUENCIAS
La principal característica de estas “ÓMICAS” es que todas ellas, unas más que otras, tienen que ver con la información extraída a gran escala o con datos generados por tecnologías/plataformas integradas de alto rendimiento. Por ejemplo, estos datos pueden constar de millones de secuencias de DNA, que constituyen piezas de un genoma, obtenidas de plataformas de secuenciación de nueva generación (NGS: next generation sequencing technology); de la intensidad de color medida por millones de sondas en un estudio de la expresión diferencial de miles de genes (Microarray expression analysis platforms), o de la detección de la presencia/ausencia de millones de SNPs o de variaciones en el número de copias en el genoma (CNVs-SNPs arrays platforms).
De igual modo, ya sean sólo algunos cientos o incluso algunos millones de datos, todos ellos son comúnmente dependientes de plataformas integradas. Por las razones anteriores, el advenimiento y crecimiento de las ciencias “ÓMICAS” está directamente relacionado con los dramáticos saltos en el desarrollo tecnológico de las plataformas de muy alto rendimiento.
Un ejemplo ilustrativo de dicho impacto puede verse reflejado per se en el Proyecto del Genoma Humano (HGP) que tomó casi diez años para completarse, utilizando las tecnologías disponibles en aquel momento, cuando se generaron los fondos gubernamentales para un consorcio internacional, cuyo objetivo era secuenciar el primer genoma humano. Podemos comparar entonces los años que tomó realizar el proyecto, contra las semanas que ahora toma secuenciar un genoma humano completo, utilizando una plataforma de nueva generación (NGS), que ocupa un espacio reducido en el laboratorio.
Lo más irónico es que este tipo de secuenciación, llamada los últimos cinco años “de nueva generación”, no es ya más lo último en tecnología, pues constantemente emergen nuevas plataformas, y ahora contamos con plataformas de tercera y cuarta generación. Por esta razón, es común que cuando los laboratorios empiezan a familiarizarse con una tecnología, haya surgido otra nueva, con muchas y nuevas ventajas, y la previa parezca obsoleta.
LIMITACIÓN COMPUTACIONAL
El primer reto que se vislumbra al contemplar estas disciplinas “ÓMICAS” es sin duda la administración y el manejo de la tremenda cantidad de datos que han sido generados. Sólo para proporcionar una idea: el reciente Proyecto de los 1000 Genomas (la secuenciación de 1000 genomas humanos) generó en sólo seis meses, más datos en el GenBank (un repositorio de DATABASE de secuencias de DNA), que los que se habían acumulado durante sus 20 años de existencia. Además de lo anterior, cada vez es más evidente la limitación en la capacidad de las computadoras para procesar esa cantidad de información y la dificultad para incrementar la velocidad de procesamiento de datos de una manera armónica.
Estas limitaciones conducen a la pregunta planteada en una edición reciente de la revista Science (Pennisi, 2011) –sobre si los equipos de cómputo podrían interrumpir el desarrollo de la genómica. Así pues, el cuello de botella en la era de las “ÓMICAS” ya no es la generación de datos, sino más bien su almacenamiento, su análisis y su interpretación. Las principales preguntas que surgen son: ¿quiénes harán y cómo harán esta tarea?
LA BIOINFORMÁTICA EN LA ERA DE LAS ‘ÓMICAS
Del mismo modo que las “ómicas”, la Bioinformática es una disciplina relativamente nueva, definida originalmente como la aplicación de la informática y la estadística a los campos de la genómica. A pesar de que creció como una herramienta para manejar la información genómica, que ahora se extiende a la mayoría de las disciplinas-ÓMICAS, el objetivo principal de la Bioinformática es facilitar y mejorar la comprensión de los procesos biológicos, a través del desarrollo de los algoritmos y métodos de cálculo y estadística.
La Bioinformática se ha convertido en el caballo de batalla del análisis de cualquier laboratorio biológico o genómico moderno, que genera sus datos en plataformas de alto rendimiento. Éste es el caso de la Unidad de Biología Molecular, Genómica y Secuenciación (UBMGyS) del Centro de Investigación y Desarrollo en Ciencias de la Salud, de la Universidad Autónoma de Nuevo León (CIDCS-UANL), que alberga su propio grupo de Bioinformática.
Esta unidad opera en la actualidad algunas de las plataformas más avanzadas tecnológicamente, como la plataforma Titanio FLX 454 (Roche) para la secuenciación de genomas, transcriptomas o metilomas completos, o las plataformas de Microarreglos (Affymetrix y Nimblegen) para la determinación del genotipo, análisis de pérdidas o ganancias de material genético y estudios masivos de la expresión génica global, que requieren del despliegue y desarrollo de complejos métodos estadísticos y de cómputo intensivo para algoritmos.
EL RETO DEL ALMACENAMIENTO E INTEGRACIÓN DE DATOS
La secuenciación del genoma completo de un organismo es en realidad mucho más compleja que alimentar al secuenciador con muestras de DNA. Secuenciadores como el Titanium 454 FLX proporcionan secuencias de ADN de tamaños cortos (llamadas lecturas), con un promedio de longitud de alrededor de 350 bases. Si comparamos este tamaño con los 4,6 millones de pares de bases del tamaño del genoma de Escherichia coli, el de 12,1 millones de bases de Sacharomices cerevisiae o los 3.200 millones de pares de bases del genoma humano, nos damos cuenta que lo que genera un secuenciador es en realidad un gigantesco rompecabezas hecho de cientos de miles a millones de lecturas, que posteriormente deben ensamblarse en forma correcta.
Generar la secuencia completa de un genoma a partir de estas lecturas implica varios pasos computacionales exigentes para la formación de “contigs” (secuencias continuas de DNA), basados en los alineamientos de múltiples lecturas para proporcionar el orden correcto de estos contigs, y la refinación del genoma ensamblado con genomas de referencia. Esto se vuelve posible gracias a los numerosos programas y equipos de computación a disposición del grupo de trabajo, tales como estaciones de trabajo y servidores (clusters).
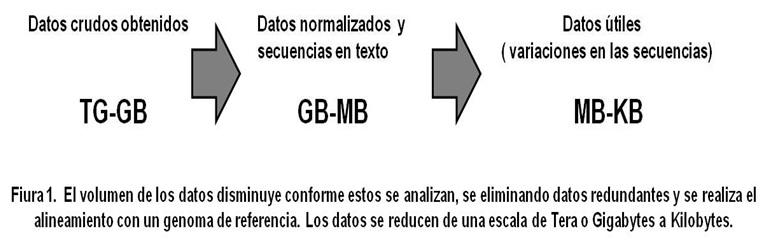
Por otro lado, ha habido un cambio dramático en el tipo de los datos que están generando las nuevas plataformas genómicas; por ejemplo, ahora se obtiene un gran número de secuencias cortas o pares de secuencias cortas, que reemplazan las lecturas relativamente largas obtenidas por la metodología tradicional de Sanger. Esto ha generado un cambio en la cantidad y en el formato de almacenamiento de los datos, y ha llevado a repensar la forma en que estos datos se manejan, se integran y se visualizan, generando a su vez retos constantes a la Bioinformática. (Figura 1).

Se estima que el costo de almacenar los gigabytes de los datos crudos producidos por una corrida de secuenciación es muchas veces mayor al costo per se de la generación de los datos, Una vez analizados los datos crudos, la información útil se reduce considerablemente (Figura 2), por lo que muchos investigadores tienen ahora la tendencia a eliminar todos los archivos crudos, una vez que éstos han sido analizados (Batley, J., 2009).

Figura 2. Debido a la Secuenciación de Nueva Generación (NGS) el costo de secuenciación por base está bajando más rápidamente que el costo del almacenamiento de un byte. [Las escalas son logarítmicas y no corregidas por los costos actuales de inflación, costos propios de laboratorio o depreciación. Imagen tomada de Baker, M. 2010].
La corrección del genoma ensamblado es esencial, ya que sirve como base para futuras investigaciones; sin embargo, es un poco problemática, como ya se mencionó. De hecho, se demostró recientemente que hasta un 20 por ciento de los genomas no humanos disponibles en las bases de datos presentan contaminación por secuencias específicas de DNA humano, incluyendo este caso de una quimera compuesta de un genoma bacteriano y el fragmento de un cromosoma humano (Longo, O.M.J Neill, y RJO Neill, 2011).
PROGRAMAS VISUALIZADORES DE GENOMAS
De la misma manera que los genomas de referencia están disponibles en las bases de datos públicas, se han desarrollado varios programas visualizadores de genomas (genome viewers) para permitir el acceso a los datos e integrarlos en forma didáctica. Los navegadores más comunes incluyen EnsEMBL, GBrowse, el de la Universidad de California, Santa Cruz Genome Browser, etcétera. Estos navegadores ofrecen a la vez varios niveles de simplicidad o sofisticación. Algunos tienen herramientas de interrogación más apropiadas que otros, dependiendo del tamaño y complejidad de los datos, así como de la necesidad del investigador, de unir o integrar la variable información colectada, tal como variación en la secuencia, comparación de genomas, identificación de marcadores genéticos, expresión diferencial de genes, estructuras de proteínas, rutas de señalización, etcétera. Estas necesidades se van censando de acuerdo a los intereses de los investigadores con la disposición de su grupo de Bioinformática.
BIOINFORMÁTICA EN EL CIDCS-UANL
Debe considerarse que, a pesar del corto tiempo (dos años) de existencia del grupo de Bioinformática, éste ya se ha integrado, ha adquirido/implementado las herramientas necesarias (tanto de softwares, estaciones de trabajo y servidores) y ha logrado participar activamente en los proyectos de investigación en Ciencias Ómicas del CIDCS-UANL. Esto es de gran importancia y trascendencia, porque marca la diferencia con respecto a otros grupos que han incursionado en las ciencias genómicas, que generan su información en plataformas cerradas y predefinidas, que les impiden analizar genomas completos, que no generan información relevante y que tienen la limitante de no poder generar nuevos descubrimientos. Nuestro grupo cuenta con amplias posibilidades y flexibilidad para la generación y análisis de nueva información.
Adicionalmente, los miembros de este grupo no se limitan al conocimiento de la biología molecular, de la genómica y la secuenciación; sino que también coordinan proyectos de investigación con otras unidades de investigación del mismo CIDCS, con distintas facultades de la Universidad, con colaboradores nacionales y con grupos internacionales de investigación.
PROYECTOS
Algunos de los proyectos en los que nuestro equipo de Bioinformática trabaja actualmente consisten:
a) En el análisis de datos de expresión obtenidos por microarreglos de diversos proyectos en cáncer (leucemias, cérvix, mama, colon, pulmón y próstata) o en diversas enfermedades complejas, como el vitíligo y labio y paladar hendido.
b) En el análisis de secuencias de genes blanco (gene targeting), para genes como BRCA1 y BRCA2, CFTR, VGFR, entre otros.
c) En el análisis de exomas (exome capturing) para Prúrigo Actínico. En estos proyectos, el objetivo es identificar biomarcadores asociados específicamente a la enfermedad, para determinar genes involucrados en la etiología de las enfermedades o que modifican el riesgo individual para estos padecimientos, o que hacen a un individuo susceptible o no a una terapia. También se trabaja en el ensamble de los genomas de organismos completos secuenciados por la UBMGyS (Figura 3).

Figura 3. Ejemplo de datos generados en el alineamiento de contigs formados con las secuencias leídas, comparadas contra una secuencia de referencia, utilizando el Software Mauve (Darling et al, 2004).
Por último, la investigación está organizada para ofrecer oportunidades a los estudiantes de licenciatura, maestría y doctorado de diversas facultades de la UANL, para poner en práctica sus habilidades y generar otras nuevas, con equipos y herramientas de clase mundial.
Bibliografía:
Baker, M. 2010. Next-generation sequencing: adjusting to data overload. Nature Methods 7, 495 – 499.
Batley, J. and Edwards D.. 2009. Genome sequence data: management, storage, and visualization. BioTechniques 46:333-336 (April 2009 Special Issue). Genome Res. 2004 Jul;14(7):1394-403.
Darling AC, Mau B, Blattner FR, Perna NT. 2004. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res ;14(7):1394-403.
Longo MS, O’Neill MJ, O’Neill RJ. 2011. Abundant human DNA contamination identified in non-primate genome databases. PLoS One. 16;6(2):e16410.
Pennisi E. 2011. Microbiology. Girth and the gut (bacteria). Science.1;332(6025):32-3.


